
Vibe Cloning


In science, there is a strong correlation between precision and cost. The more precisely one measures something, the more the measurement typically costs. A low-resolution light microscope costs about $500. A confocal microscope, capable of resolving condensed chromosomes or individual actin filaments, costs about $250,000.
Most scientists consider this a worthwhile tradeoff. After all, science is about devising a more complete description of the universe, and making more precise measurements is a big part of that.
But there are many cases in which less precision is actually a good thing, especially as experiments scale. A high experimental cost doesn’t matter much when scientists run an experiment one time, but higher costs quickly become a bottleneck when they repeat it thousands of times.
If you buy into the thesis that AI models will soon propose original research questions, then you probably also believe that the bottleneck in testing those ideas will, increasingly, shift toward the real-world. The cost and speed of experiments will be the major bottleneck for collecting wet-lab data to improve or benchmark AI models. The difference between sequencing a gene for $10 vs. $20 may seem trivial, but becomes a big deal at the scales biologists will soon need to think about.
Scientists should, therefore, embrace “vibe cloning,” or the idea that sequence perfection does not matter in cases where gene mutations are unlikely to impact function. In fact, a big opportunity exists for philanthropists to fund a ~million dollar effort toward a tolerance model, or computational tool which predicts which sequence variations are permissible across proteins. Doing so would not only help us understand biology more deeply, but also save money on experiments performed at scale.
This may seem foolish, but we think the high precision our experiments require is actually causing us, collectively, to misunderstand how biological systems work1. Scientists often describe DNA as a code, leading to an implicit assumption that biological systems require digital-grade fidelity to function. But they definitely do not. In AI, “slop” is perceived as low quality; a failure mode and something to be filtered out. In biology, though, slop is the mechanism by which evolution creates new functions. Imperfect DNA replication, point mutations, and recombination errors are all natural parts of adaptation.
To understand this more clearly, it probably helps to give an example. So let’s talk about DNA cloning, a ubiquitous method in biotechnology.
Mutations are Rare and Mostly OK
Thousands of scientists clone genes every day. And yet the technique, despite being nearly half a century old, remains incredibly tedious and expensive.
Say you want to clone a gene encoding green fluorescent protein, or GFP, into E. coli. Specifically, the goal is to splice (or insert) this gene into a plasmid, or loop of DNA, that the bacteria will “read” and express, thus glowing a fluorescent green.
Cloning begins by first isolating an existing GFP gene and making lots of copies of it. You can take a vial of DNA from a labmate and then use the polymerase chain reaction, or PCR, to make billions of copies.
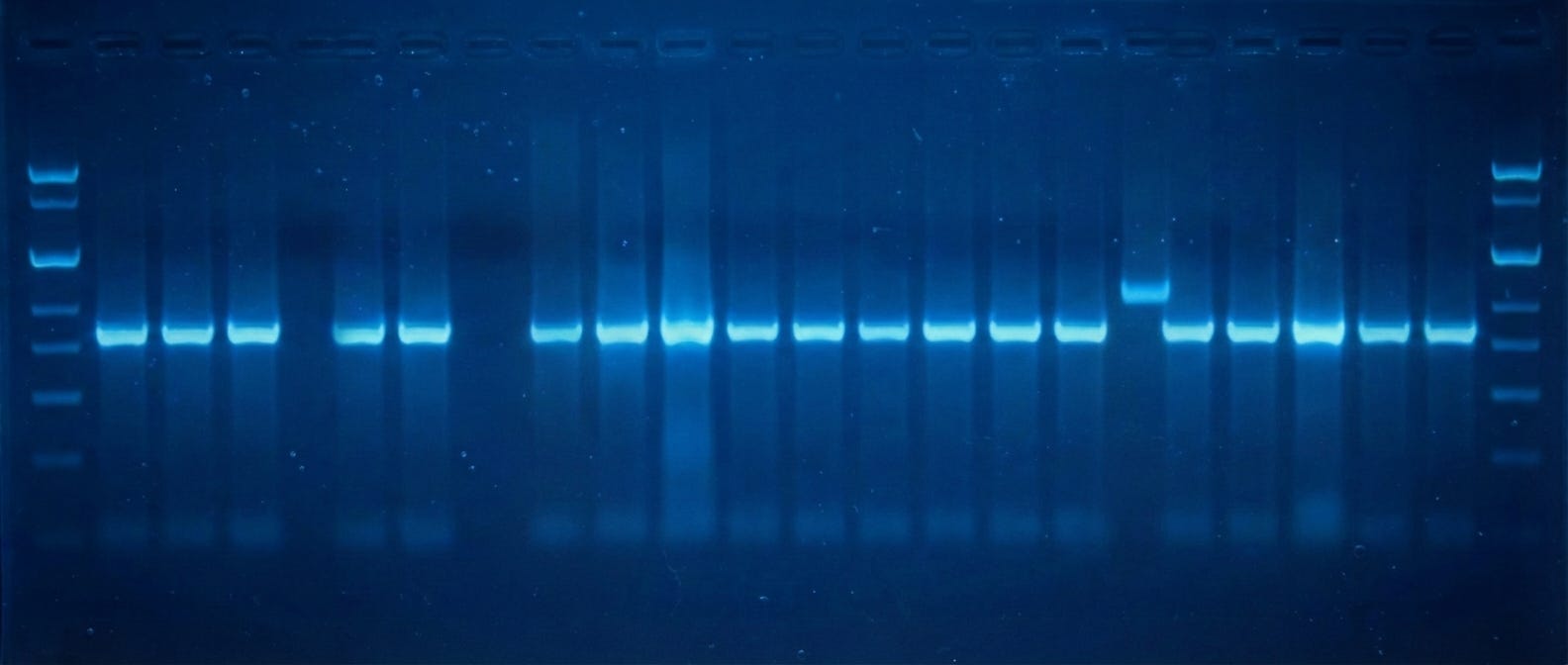
Next, you use restriction enzymes to cut both the amplified GFP genes and the plasmid into which GFP will be pasted. These enzymes snip the DNA and leave behind “overhangs” that enable the two pieces to be joined together later (using yet another enzyme, called a ligase.) But first, both fragments are run on an agarose gel. An electric current pushes the DNA strands through the gel, with smaller pieces moving faster. By running the snipped DNA alongside a reference standard, or “ladder,” you can figure out the size of each piece. A DNA stain makes the bands visible.
Scientists then cut out the bands corresponding to GFP and the plasmid from the gel with a scalpel, purify them, and ligate them together. They transform this ligation mixture into E. coli cells and plate them onto agar plates spiked with antibiotics. After at least eight hours, colonies become visible and — ideally — glow green, indicating both that the cells have an antibiotic resistance gene (encoded on the plasmid) and encode GFP.
Scientists pick these colonies with a plastic or metal loop, grow them in broth for several more hours, and then isolate the plasmids and send them for sequencing. Scientists usually pick 5–10 colonies just to make sure they find one that has exactly the correct sequence. Companies like Plasmidsaurus will sequence an entire plasmid for $15, so if you pick 10 colonies, that costs $150 total. (This seems small. But consider that Addgene catalogs ~170,000 plasmids, at an estimated sequencing cost of about $25M excluding time and labor.)
Sequencing is both the most expensive and most time-consuming part of DNA cloning. So what would it look like to cut this by 90 percent or more? What would it mean to pick just one colony, accept mutations, and move on?
Everything would probably be fine, for two reasons. First, because mutations are unlikely. And second, because biology is highly tolerant of variations.
In fact, we can calculate the likelihood that a plasmid will gain a mutation during cloning. E. coli bacteria accumulate about 1.5 × 10⁻³ mutations per genome per generation, according to a 2015 study. That is across the entire genome; all 4.6 million base pairs. A plasmid with 3,000 nucleotides, for example, is a much smaller target. But given this baseline mutation rate, the probability that the plasmid will collect at least one mutation in a given generation is 1 in 1.7 million. Further, if we assume that cells go through 30 generations during overnight growth (one of the last steps of cloning), then about 50,000 cells out of every one billion E. coli (or just 0.005% of the population) will have a plasmid carrying a mutation.
Most plasmid mutations arise during DNA cloning from the PCR step instead. Taq polymerase has an error rate of about 8 × 10⁻⁶ per bp per PCR cycle. So if your insert has 3,000 nucleotides, and you amplify it over 25 cycles of PCR, then about 45% of copies will have at least one error by the time you’re finished. This is a lot, but a more accurate polymerase, like Phusion, reduces this to about 3% of copies.
Even if the cloned plasmid does have a mutation, it’s likely a benign change. Most mutations have no effect on function. A missense mutation in a loop region probably won’t affect anything, and most substitutions do not meaningfully alter the brightness or folding of the protein.
Experimental evidence supports this claim. For a 2023 paper, researchers did the (unenviable) task of designing and screening a huge library of GFP variants, ultimately finding 16,000 unique variants that still fluoresced. Some of these variants had as many as eight mutations in their active sites, or part of the protein that is actually responsible for the glow. In other words, the most common errors from synthesis — like single-base substitutions — are the most likely to be functionally permissible. And the catastrophic errors, like frameshifts or large deletions, can easily be caught using functional screening instead of sequencing.
The central thesis of vibe cloning, then, is that we should stop obsessing over sequence perfection in cases where gene mutations are unlikely to impact function. If we pick two colonies after cloning instead of ten, we will save $120 each time. This adds up over a year, or over a PhD, or across thousands of researchers.
Instead of sequencing ten colonies to find a perfect match, we should instead test whether each construct works using a three-tiered approach:
Selection: if a construct carries a resistance marker and the host survives, the plasmid is intact.
Expression: use a fluorescent reporter or other assay to confirm the construct makes protein at the colony level.
Activity: test whether the construct does what you actually want by measuring enzyme activity, growth curves, or something else.
The cell is the compiler, and the compiler has been more forgiving than we have given it credit for.
Of course, there are problems with “vibe cloning.” For one, it doesn’t work with library-based approaches, where many constructs are tested in a pool, because those pools cannot easily be assessed for function. If the expressed gene imposes a fitness cost on the host, broken constructs outcompete functional ones in bulk selection.
There are other objections to our arguments, too. Among them, surely, is: “But what about reproducibility? If we allow random mutations into a plasmid, how will other laboratories replicate the work?”
Fair point! But the solution is easy; just report the sequence in the final paper, regardless of what it is. If your GFP carries a Val→Ile substitution at position 117, then just write that down in the methods section or supplemental materials. Reproducibility does not require that every lab on Earth uses an identical copy of the same gene, but rather that every lab knows what gene was used.
Another problem with “vibe cloning” is that we cannot always know, de novo, which mutations are likely to matter. There is always a non-zero chance that a mutation will cause a protein to misfold or break. So if we just accept any gene sequence, some experiments will fail for unexpected reasons. This is a real concern, but not unsolvable.
Tolerance Models
The solution is to build a tolerance model, or framework that tells us which sequence variations are permissible for which genes and functions. What’s remarkable is that every molecular biology lab is already generating the data needed to build this model, yet it’s being thrown away.
Today, when a scientist sequences a clone and finds a point mutation, that clone gets thrown away. Nobody even writes down what the mutation was, where it occurred, or whether the construct might have worked anyway.
The opposite is also true. Imagine a scientist picks a colony, skips sequencing because they are in a hurry, tests the construct directly, and it works. The construct carries whatever errors synthesis and assembly introduced, yet is functional despite them. But because the scientist never sequenced it, the specific variations it carried are unknown, and the observation that it tolerated those variations is never recorded.
Scientists should collect both halves of this tolerance dataset — sequences that deviated and failed, as well as sequences that deviated and worked — and use them to train a predictive model.
Such a model is valuable for two reasons. First, because it would save enormous amounts of money and time. If we know that GFP tolerates mutations at 80% of its positions, we can reduce sequencing from ten colonies to two or three, collectively saving millions of dollars each year. And second, because we would learn something fundamental about biology itself; which positions in proteins are critical or flexible, about the relationship between sequence and function, and about the tolerance of biological systems to variation.
We should start with the genes that appear most frequently in cloning experiments, like GFP, RFP, antibiotic resistance markers, and promoters. The 2023 study that screened 16,000 GFP variants did this for one gene, but now we should repeat it systematically for the entire molecular biology toolkit. The dream would be to have a “Vibe Cloning” portal that curates these data and uses an AI model to predict whether a mutation is acceptable or not.
This idea may seem outlandish, but Cultivarium is already working on it! The speed and cost of experiments is, after all, the greatest bottleneck to biological progress, because it fundamentally limits the number of ideas we can test. Therefore, we ought to find more places where we can relax precision in favor of scale.
Most biology research isn’t reproducible anyway. Large-scale replication studies have found that somewhere between a third and two-thirds of published results do not hold up when other groups try to repeat them. Most methods are also poorly described (to be charitable), and even the quality of deionized water can often determine whether or not an experiment works. Relaxing the precision of our gene sequences, surely, should not be the top concern in metascience!
 | A guest post by
|
FWIW we did this back in my day at Ginkgo - especially if you always sequence winners. Often stuff coming out of synthesis was sequenced by default - if some library members weren’t perfect we’d log the mutations and move them forward anyway rather than waiting for another round of synthesis just for a few stragglers. Lots of 80/20 decisions in keeping a project moving forward quickly
A salty/speculative riff here could be that molecular biology was founded by physicists - we needed their incredible rigor to understand the systems with the tiny little light that we were able to shine on it. That was the “I don’t believe a word of it!” Days led by Max Delbruck et al
But this essay pushes on the idea that our limitation is different today, so the leading mindset is different. “Extreme throughput over extreme precision” def shoves out the physicists and welcome the engineers